|
West-Syndrom
Das West-Syndrom ist eine vergleichsweise seltene und schwer zu behandelnde Form generalisierter maligner Epilepsie.
Sie ist altersgebunden, tritt bei Säuglingen in der Regel in der Zeit zwischen dem dritten und zwölften Monat nach der Geburt auf und erreicht den Manifestationsgipfel durchschnittlich im fünften Monat. Die Ursachen können vielfältig sein (Polyätiologie); häufig liegt dem Syndrom eine tief greifende hirnorganische Störung zugrunde, die entweder vorgeburtlich (pränatal), während der Geburt (perinatal) oder nachgeburtlich (postnatal) entstanden ist.
Als Synonyme für den Ausdruck West-Syndrom werden die Begriffe maligne Säuglingsepilepsie, infantile Spasmen, Propulsiv-Petit-mal und im deutschsprachigen Raum BNS-Epilepsie als Abkürzung für Blitz-Nick-Salaam-Epilepsie (siehe Erscheinungsbild) verwendet.
Geschichte
Das West-Syndrom wurde nach dem englischen Arzt und Chirurgie|Chirurgen William James West (1793-1848) benannt, der in Tonbridge (Grafschaft Kent) lebte. Er beobachtete diese besondere Form der Epilepsie im Jahre 1841 bei seinem eigenen, damals etwa vier Monate alten Sohn und veröffentlichte seine Beobachtungen unter wissenschaftlichen Gesichtspunkten in einem Artikel im The Lancet. Er bezeichnete die Krämpfe damals als Salaam-Tic.
Häufigkeit
Die allgemeine Auftrittswahrscheinlichkeit (Prävalenz) liegt bei etwa 1:4000 bis 1:6000, Statistik|statistisch gesehen sind Jungen im Verhältnis von etwa 3:2 häufiger vom West-Syndrom betroffen als Mädchen. Bei etwa 45 von 50 der Kindern treten die Anfälle erstmals in der Zeitspanne vom dritten bis zum zwölften Monat nach der Geburt auf. In selteneren Fällen setzen die Anfälle in den ersten beiden Monaten oder im Verlauf des zweiten bis vierten Lebensjahres ein.
Ursachen
Welche Biochemie|biochemischen Mechanismen zum Auftreten eines West-Syndroms führen, ist bislang nicht bekannt. Vermutet wird eine Störung der Neurotransmitterfunktion, genauer gesagt eine Störung der Regulation des GABA-Stoffwechsels. Eine andere Möglichkeit, die erforscht wird, ist eine Überproduktion des Corticotropin Releasing Hormones. Beide Hypothesen (denkbar wäre auch ein multifaktorielles Zusammenspiel) werden gestützt durch die Wirkungsweise verschiedener Medikamente, die zur Behandlung des West-Syndroms eingesetzt werden.
Als mögliche Ursachen werden erwogen:
- Bei etwa zwei Drittel der Kinder ist eine tief greifende hirnorganische Störung nachweisbar. Dazu zählen z. B. Mikrozephalie, Entwicklungsstörungen der Hirnrinde (''Cortikale Dysplasien''), Hirnatrophie, Lissenzephalie (Agyrie oder Pachygyrie), Gehirnentzündung, Bakterien|bakterielle Meningitis, Phakomatosen (z. B. tuberöse Sklerose / Bourneville-Syndrom), Aicardi-Syndrom, subdurales Hämatom und Fehlbildungen von Blutgefäßen (vaskuläre Malformationen).
- Des Weiteren werden in der Literatur als Ursache vermehrt neurometabolische Erkrankungen, angeborene Infektionen (z. B. Cytomegalie), Hypoglykämie, Hirnschäden durch Asphyxie oder Hypoxie (Sauerstoffmangel, z. B. unter der Geburt, hypoxisch-ischämische Enzephalopathie), periventrikuläre Leukomalazie, Hirnblutungen, Schlaganfall oder Schädel-Hirn-Traumata verschiedener Art, sowie Schädigungen des Gehirns durch Frühgeburtlichkeit genannt.
- Es sind Fälle bekannt, in denen die Krämpfe als mögliche Nebenwirkung erstmalig nach einer Kombinationsimpfung (Mehrfachimpfung) gegen Masern, Mumps und Röteln oder gegen Tetanus, Pertussis, Diphtherie, Poliomeyelitis, Hebatitis B und Haemophilus influenzae Typ b-Erkrankungen auftraten. Jedoch ist die Herstellung eines Zusammenhangs zwischen Impfung und West-Syndrom schwierig, da vielfach die Impfung in die Zeit fällt, in der die Anfälle auch sonst typischerweise erstmals auffällig werden. Anerkannt ist das West-Syndrom als Impfschaden nicht.
Lässt sich eine Ursache nachweisen, spricht man von einem ''symptomatischen West-Syndrom'', da die Anfälle als Begleiterscheinung oder Merkmal (Symptom) einer anderen Besonderheit auftreten.
Von einem ''kryptogenen West-Syndrom'' spricht man gelegentlich, wenn ein symptomatisches West-Syndrom zu vermuten, nicht aber nachgewiesen ist. Bei etwa 20% der betroffenen Kinder lässt sich keine Ursache für die Krämpfe finden. Wichtige Diagnosekriterien sind: Eine bis zum Auftreten der Anfälle oder vor Beginn der Therapie regelgerechte Entwicklung mit unauffälligen neurologischen und neuroradiologischen Befunden sowie kein Nachweis einer auslösenden Ursache für die Krämpfe.
In etwa 15% der Fälle bekommen innerhalb der Familie mehrere Kinder das West-Syndrom. In diesem Fall spricht man von einem ''idiopathischen West-Syndrom'', bei dem Genetik|genetische und mitunter erbliche Einflüsse eine Rolle spielen. Es sind Fälle bekannt, in denen das West-Syndrom in aufeinander folgenden Generationen bei Jungen auftrat; es handelt sich dabei um einen x-chromosomalen Erbgang (Genort Xp22.13 / ARX, aristaless, Homöobox-Gen).
Erscheinungsbild
Die epileptischen Anfälle, die bei Säuglingen mit West-Syndrom beobachtet werden können, lassen sich in drei Anfallsformen gliedern, die der Besonderheit das Synonym BNS-Epilepsie eingebracht haben.
Meist gleichzeitig, zum Teil jedoch auch unabhängig voneinander, tritt beim West-Syndrom in typischer Form folgende Trias von Anfallstypen auf:
- Blitz-Anfälle (B): Plötzlich (blitzartig) auftretende heftige myoklonische Zuckungen des gesamten Körpers oder einzelner Körperteile in Sekundenbruchteilen, wobei in der Regel insbesondere die Beugung der Beine auffällig ist (Beugemuster sind hier generell häufiger als Streckmuster).
- Nick-Anfälle (N): Zuckungen der Nacken- und Halsmuskulatur, wobei das Kinn ruckartig in Richtung Brust gebeugt oder der Kopf eingezogen wird (nicken).
- Salaam-Anfälle (S): Schnelle Beugung des Kopfes und des Rumpfes nach vorne und gleichzeitiges Hochwerfen und Beugen der Arme mit teilweisem Zusammenführung der Hände vor der Brust und / oder Ruderbewegungen. Würde man sich diesen Vorgang verlangsamt vorstellen, ähneln die Bewegungen dem morgenländisch-orientalischen Friedensgruß (Salaam), was diesem Anfallstyp den Namen eingebracht hat.
Die Anfälle treten unabhängig von äußeren Reizen auf und können häufig kurz nach dem Aufwachen oder kurz vor dem Einschlafen bei den Kindern beobachtet werden. Sie können jedoch auch zu anderen Zeiten auftreten und z. B. auch im Schlaf beginnen und zum Aufwachen führen. Manchmal treten sie anfangs vereinzelt, später dann in Serien (''Clustern'') von bis zu 150 Anfällen auf, wobei die Intervall Intervalle zwischen den Krämpfen jeweils weniger als 60 Sekunden betragen.
Obgleich die Anfälle nicht mit Schmerzen verbunden sind und das Bewusstsein vermutlich erhalten bleibt, weinen die Kinder häufig während der Anfälle oder oft auch danach, da sie sehr anstrengend sind. Da diese Form der Epilepsie selten und daher wenig bekannt ist, werden die BNS-Anfälle von vielen Eltern zunächst als Schreckreaktion, als Moro-Reflex (Umklammerungs-Reflex) oder als Blähungen und Bauchschmerzen gedeutet; letzteres insbesondere auch aufgrund des Weinens der Kinder. Oft holen sie erst dann ärztlichen Rat ein, wenn die Anfälle ihres Kindes in Serien auftreten und das unübliche Bewegungsmuster dadurch deutlich wird.
Sonstige Merkmale, die auffallend häufig bei Kindern mit West-Syndrom beobachtet werden können (oft auch schon bevor die Anfälle einsetzen), sind z. B.:
- bei 45 von 50 Kindern: allgemeine psychomotorische Entwicklungsverzögerung (ggf. Rückschritte oder Stillstände in der bereits erfolgten Entwicklung)
- gestörte Kontaktaufnahmefähigkeit, oft mit gestörtem Blickkontakt, Verlernen des (sozialen) Lachens
- unübliche Augenbewegungen (häufig: Abweichung der Augenachse mit kurzen Blicken nach oben, Augenzittern)
- Schwerhörigkeit (keine üblichen Reaktionen auf Geräusche und Ansprache)
- Muskelhypotonie (Verringerung der Muskelspannung; oft erkennbar daran, dass die Kinder ihren Kopf nicht altersentsprechend halten können.)
- Grimassieren (teils mit unüblichem Schmatzen oder Gähnen)
- Stimmungsschwankungen, Nachlassen der Vigilanz bis hier zur Apathie einerseits und zeitweise besondere Unruhe andererseits
- weiße Flecken auf der Haut
Nicht alle Merkmale kommen bei allen Kindern vor oder sind in gleich starker Ausprägung nachweisbar.
Manchmal sind abgeschwächte Anfallstypen zu beobachten, die (zunächst) nicht dem klassischen Bild eines BNS-Anfalls entsprechen (z. B. gesondertes Augenverdrehen, Kopfdrehungen, einseitige Extremitätenbewegungen) oder mit anderen Anfallsformen (z. B. Grand-Mal-Anfällen) einhergehen. Die Krämpfe beim West-Syndrom können auch beim einzelnen Kind hinsichtlich ihrer Intensität und Länge sehr variabel sein. Das Spektrum reicht von unscheinbaren Zuckungen bis hin zu ausladenden Bewegungsfolgen, die den gesamten Körper betreffen. Die Dauer beträgt zwischen wenigen Sekunden und mehreren Minuten, manchmal entwickelt sich sogar ein Status epilepticus. Beim klassischen Verlauf zeigen sich zunächst meist wenige schwache Verkrampfungen von kurzer Dauer, die sich nach und nach zu Serien häufen.
Etwa 15 von 50 Kindern mit West-Syndrom haben eine zusätzliche konstitutionelle Anfallsbereitschaft.
Auswirkungen
Obwohl die Anfälle aufgrund ihrer Kürze und Unscheinbarkeit für Laien harmlos erscheinen mögen, führen sie unbehandelt oder bei Therapieresistenz zu schweren und zum Teil dauerhaften Störungen der kognitive Behinderung|kognitiven und körperlichen Entwicklung des Kindes.
Konzentrationsdefizite aufgrund der bestehenden epileptischen Überaktivität im Gehirn können zu Lernschwierigkeiten führen, bereits erworbene Fähigkeiten wie der lautsprachliche Ausdruck und das soziale Lachen werden nicht selten wieder verlernt. Eine nachlassende Muskelspannung (Muskelhypotonie) schränkt die Bewegungsfähigkeit ein. Häufig wird ein gestörtes Spielverhalten beobachtet, Autismus|autistische Züge sind möglich.
Diagnose
Kinder mit West-Syndrom fallen häufig bereits vor dem erstmaligen Auftreten der Anfälle durch eine nicht altersentsprechende Entwicklung auf.
Das West-Syndrom als Epilepsie mit generalisierten Anfällen fokal und multifokal gelagerter Entstehungsregion manifestiert sich mit einer sogenannten Hypsarrythmie: Diagnostische Relevanz hat das typische EEG-Muster, bei dem sich die epileptische Aktivität in Form unregelmäßig hoher Deltawellen mit Synchronität|desynchron auftretender Einstreuung von kurzdauernden Spitzenpotentialen (Spikes) und / oder Sharp waves zeigt. Dauer und Lokalisation (fokal/multifokal) sind dabei unsymmetrisch, Variationen sind die Regel: "Die Hypsarrythmie tritt nie als rhythmisches und gut organisiertes Muster auf" (Gibbs & Gibbs, 1952).
Auch das Bild einer sogenannten "modifizierten Hypsarrythmie" mit einzelnen beidseitig einheitlichen Entladungen ist beschrieben worden, sowie die Variante der "Hemihypsarrythmie'', die auf eine einseitige Hirnschädigung zurückzuführen ist.
Zwischen den Anfällen und manchmal auch nur im Schlaf zeigt sich ein Bild langsamer Wellen und beidseits hoher Spitzen.
Neben der Diagnose durch die Messung der elektrischen Aktivität im Gehirn des Kindes wird in der Regel empfohlen, eine Analyse des Blutes und des Urins vornehmen zu lassen, um das Vorliegen von Chromosomenbesonderheiten, Erbkrankheiten, Stoffwechselkrankheiten (z. B. Phenylketonurie) und Infektionskrankheiten zu überprüfen, um hier ggf. behandelnd eingreifen zu können und um die Ursache der epileptischen Anfälle zu klassifizieren bzw. einzugrenzen.
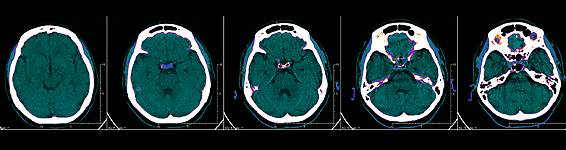
Die Überprüfung des Vorliegens einer hirnorganischen Besonderheit ist durch eine bildgebende Untersuchung des Gehirns möglich. Gängige Verfahren sind:
- Ultraschall (bei Säuglingen machbar, da sich das knöcherne Schädeldach noch nicht geschlossen hat)
- Magnetresonanztomografie (MRT ): Überprüfung auf Hirnfehlbildungen, Hirnstamm-Atrophie, Verzögerung der Myelinisierung
- Positronenemissiontomographie (PET): Überprüfung auf kortikale Dysplasien bei lokalem Glukose-Hypometabolismus, Hypermetabolismus der Linsenkerne)
- Single-Photon-Emissionstomographie (SPECT): Überprüfung auf lokale Durchblutungsstörungen
- Computertomographie (CT): Überprüfung auf Hirnfehlbildungen, Kalzifizierungen, Atrophien.
Treten die Bewegungsmuster der Anfälle seitenbetont auf, lässt dies eine Hirnschädigung der entsprechenden Seite vermuten, der nachgegangen werden sollte. Häufig finden sich z. B. Erweiterungen der Hirnventrikel (Hirnwasserräume), narbenartige Verdichtungen, Verkalkungen oder knotige Besonderheiten des Hirngewebes, Besonderheiten der Hirnfurchung oder eine nicht altersentsprechende Hirnreifung.
Allgemeinmedizinisch orientierte Kinderärzte besitzen oft wenig Erfahrung mit dem im Vergleich zu anderen Epilepsien seltenen West-Syndrom und dessen Symptomatik, was zu Fehldiagnosen wie Moro-Reflex (Umklammerungs-Reflex), Bauchschmerzen oder Blähungen führen kann. Ein Kind mit der Verdachtsdiagnose West-Syndrom sollte daher auch von einem Kinderneurologen (Neuropädiater) untersucht werden. Bei Säuglingen und Kleinkindern mit unklaren Entwicklungsstörungen muss auch an das West-Syndrom und das Lennox-Gastaut-Syndrom gedacht werden.
Differentialdiagnostisch ist das West-Syndrom darüber hinaus durch den Nachweis der charakteristischen Hypsarrythmie abzugrenzen vom rhythmischen oder arhythmischen Schlaf- oder Wachmyoklonus, dem benignen (gutartigen) Myoklonus des Säuglingsalters, dem Ohtahara-Syndrom, vom Lennox-Gastaut-Syndrom, von der myoklonischen Frühenzehalopathie, von der benignen und von der malignen myoklonischen Epilepsie des Säuglings- und Kleinkindalters, sowie von Störungen, die von den Bewegungsabläufen her ein den BNS-Krämpfen ähnliches Bild zeigen können.
Therapie
Das West-Syndrom ist im Vergleich zu anderen Epilepsien schwer erfolgreich zu behandeln. Eine möglichst frühzeitige Diagnose und ein umgehender Behandlungsbeginn sind ausgesprochen wichtig, können jedoch einen Therapieerfolg nicht garantieren. Es ist noch nicht hinreichend erforscht, ob die Form der Behandlung Einfluss auf die Langzeitprognose nehmen kann. Die Prognose ist zwar nach heutigem Wissensstand überwiegend bestimmt durch die Ursache der Anfälle, allgemein kann jedoch festgestellt werden, dass ein schlechtes Ansprechen auf die Therapie (und damit einhergehend die weiterhin bestehende epileptische Überaktivität im Gehirn) eine schlechte Prognose (mit)bedingt. Die Behandlung erfolgt stets individuell und richtet sich insbesondere nach der Ursache des West-Syndroms (ätiologische Klassifikation) und der Gehirnentwicklung (Zeitpunkt der Hirnschädigung).
Ist eine behandelbare hirnorganische Besonderheit Ursache der Anfälle, ist in einigen Fällen nach gründlicher Abwägung der Vor- und Nachteile eine operative Korrektur durch Epilepsie-Chirurgie möglich. Hierdurch können die Ursache umd damit die Anfälle beseitigt werden. In den meisten Fällen des West-Syndroms basiert die Therapie jedoch auf der Gabe von Medikamenten, wobei zunächst meist versucht wird, die Anfälle durch die hochdosierte Gabe von Gammaglobulin (anfallsfrei werden zwei von zehn Kindern) oder des Vitamins B6 (Pyridoxin/anfallsfrei werden etwa drei von zehn Kindern) in den Griff zu bekommen, da in eher seltenen Fällen ein entsprechender Mangel oder eine Verwertungsstörung für die Epilepsie verantwortlich ist.
Gelingt dies nicht, wird mit der Gabe von Medikamenten aus der Antikonvulsivum-Gruppe begonnen. Es wird hierbei meist versucht, zunächst mit möglichst gut verträglichen und mit wenig Nebenwirkungen einhergehenden Präparaten oder Dosierungen zu beginnen und die Medikation ggf. zu steigern bzw. zu verändern, wenn nach einem entsprechenden Kontrollzeitraum die Anfälle nicht weniger werden oder aufhören. Nicht selten zeigen sich zudem insbesondere bei der Notfallmedikation paradoxe Reaktionen auf die Wirkstoffe (z. B. aufputschende Wirkung statt beruhigende), was manchmal sogar anfallsfördernd sein kann. Eine Umstellung sollte dann in Betracht gezogen werden. Auch unabhängig davon sind mehrmalige Medikamentenumstellungen bei der Behandlung des West-Syndroms keine Seltenheit: Je nach dem wie sich die Anfälle entwickeln wird die Dosis angepasst oder das Medikament abgesetzt und auf ein anderes Präparat umgestellt. Je nach dem Zeitraum der Gabe wird ein Medikamentenwechsel durch Ausschleichen vorbereitet, um Entzugserscheinungen zu vermeiden.
Gängige Medikamente, die zur Behandlung des West-Syndroms eingesetzt werden sind z. B.:
- Valproat (''Ergenyl®'', ''Orfiril®''): Anfallsfrei werden bis zu acht von zehn Kindern, die Rückfallquote liegt bei bis zu 30%. Nebenwirkungen sind z. B. Ernährungsprobleme und Sedierung, seltener Hepatopathie (Schädigung der Leber).
- Vigabatrin (''Sabril®''): Oft eingesetzt beim ansonsten therapieresistenten West-Syndrom und als Mittel der ersten Wahl häufig bei solchen Kindern, bei denen das West-Syndrom symptomatisch bei tuberöser Sklerose auftritt, weil es sich hier als besonders wirksam erwiesen hat.
- Topiramat (''Topamax®'')
- Sultiam (''Ospolot®'')
- Felbamat (''Taloxa®''): Oft eingesetzt beim ansonsten therapieresistenten West-Syndrom.
- Benzodiazepine
- ACTH (''Synacten®''): Anfallsfrei werden nach kurzer bis mittellanger Therapie bis zu acht von zehn Kindern (offenbar deutlich geringere Wirksamkeit bei frühgeborenen Kindern, vermutlich aufgrund des Entwicklungsstandes des Gehirns bzw. der perivaskulären weißen Substanz). Die Rückfallquote liegt bei bis zu 65%. Die Behandlung ist mit hohen Risiken durch mitunter massive Nebenwirkungen belastet, die von Dauer und Dosis abhängen (u. a. Leukozytose, Schwächung des Immunsystems, Hyperglykämie, Bluthochdruck, Erbrechen, Magenblutungen, Herzversagen, Cushing-Syndrom).
- Glukokortikoide (Nebennierenhormone): Dexamethason oder Prednisolon als Alternativmedikamente zu ACTH.
Als Notfallmedikation bei häufigen BNS-Anfällen wird oft Chloralhydrat (''Chloraldurat®''), Lorazepam (''Tavor®''), Phenobarbital (''Luminal®'') oder Diazepam verschrieben, um Cluster von mehreren Minuten Länge zu unterbrechen und einer neuen Anfallsserie vorzubeugen.
Zum Teil sind die Nebenwirkungen der Medikamente sehr belastend für den kindlichen Organismus, so dass die Kinder sich wie in einem Dämmerzustand befinden. Auch bei der Medikamentenauswahl sind die Risiken durch Nebenwirkungen und mögliche Abhängigkeit mit den gesundheitlichen Risiken durch die Anfälle abzuwägen. Viele Medikamente machen vergleichsweise schnell abhängig, so dass bei Medikamentenumstellungen oder nach dem Absetzen eines entsprechenden Präparates behandlungsbedürftige Entzugserscheinungen auftreten können.
Die Kontrolle und Dokumentation des Behandlungs- und Entwicklungsverlaufs erfolgt insbesondere durch regelmäßige Überprüfung des EEG-Bildes und bei medikamentöser Einstellung durch Blutuntersuchungen zur Überwachung des Medikamentenspiegels.
Um einer Verzögerung der körperlichen und kognitiven Entwicklung des Kindes entgegen zu wirken bzw. Entwicklungsrückschritten und -stillständen möglichst vorzubeugen, werden Maßnahmen wie z. B. Frühförderung, Motopädie, Physiotherapie, Ergotherapie und Logopädie durchgeführt. Eine individuelle, intensive und kontinuierliche Betreuung ist hierbei wichtig.
Prognose
Allgemeine Entwicklungsprognosen sind aufgrund der angesprochenen Variabilität der Ursachen und der Ausprägung der Symptomatik und Ätiologie nicht pauschal möglich. Es muss stets der Einzelfall betrachtet werden.
Bei Kindern mit kryptogenem West-Syndrom sind die Prognosen meist günstiger als bei solchen mit der idiopathischen und symptomatischen Form: Sie zeigen seltener bereits vor dem Auftreten der Anfälle Entwicklungsstörungen und neurologische Auffälligkeiten, die Anfälle lassen sich oft schneller und effektiver behandeln, die Rückfallrate ist niedriger. Die Kinder entwickeln im Anschluss an das West-Syndrom seltener andere Formen der Epilepsie und bei durchschnittlich etwa zwei von fünf Kindern zeigt sich im weiteren Verlauf eine altersgerechte Entwicklung.
Ansonsten ist die Behandlung des West-Syndroms jedoch vergleichsweise schwierig und der Therapieerfolg oftmals unbefriedigend, sodass davon ausgegangen werden muss, dass Kinder mit symptomatischem und idiopathischem West-Syndrom eine eher ungünstige Prognose haben (insbesondere bei Theapieresistenzen).
Statistisch gesehen überleben 5 von 100 Kindern mit West-Syndrom die ersten fünf Jahre ihres Lebens nicht (teils durch die ätiologische Ursache des Syndroms, teils durch medikamentenbedingte Mortalität). Nur bei weniger als der Hälfte der Kinder gelingt die Herstellung von Anfallsfreiheit durch medikamentöse Behandlung. Statistisch gesehen können etwa drei von zehn Kindern befriedigend behandelt werden, wobei sich durchschnittlich lediglich eines von 25 Kindern kognitiv und motorisch weitgehend regelgerecht entwickeln.
Ein großer Teil (bis zu 90%) der Kinder ist auch nach erfolgreicher Behandlung der Anfälle körperlich und kognitiv deutlich beeinträchtigt. Dies ist meist jedoch nicht in erster Linie auf die epileptischen Anfälle sondern auf deren Ursache (hirnorganische Besonderheit bzw. dessen Lokalisation und Schweregrad) zurückzuführen, wobei durch schwere und häufige Anfälle das Gehirn (zusätzlich) Schaden nehmen kann.
Bleibende Schädigungen, die in der Literatur mit dem West-Syndrom in Zusammenhang gebracht werden, sind neben kognitive Behinderung|kognitiver Behinderung, Lernstörungen und Verhaltensauffälligkeiten eine Cerebralparese (bei bis zu 5 von 10 Kindern), psychische Störungen, häufig Autismus (bei etwa 3 von 10 Kindern). Auch hier muss jedoch stets die individuelle Ätiologie des West-Syndroms in die Erörterung des jeweiligen Ursache-Wirkung-Komplexes einbezogen werden.
Bis zu 6 von 10 Kinder mit West-Syndrom bekommen im Laufe ihres Lebens eine Nachfolgeepilepsie: Teils geht das West-Symdrom in eine fokale oder eine andere generalisierte Epilepsie über und etwa die Hälfte der Kinder bekommt das Lennox-Gastaut-Syndrom.
West-Syndrom bei Säuglingen mit Down-Syndrom
Bei durchschnittlich 1 bis 5 von 100 Kindern mit Down-Syndrom (Trisomie 21) tritt im Säuglingsalter das West-Syndrom auf. Während diese Epilepsieform bei den meisten Kindern ohne die dem Syndrom zugrundeliegende Chromosomenbesonderheit vergleichsweise schwer erfolgreich zu behandeln ist, kann bei Kindern mit Down-Syndrom vielfach ein deutlich milderer Verlauf und eine bessere Ansprechbarkeit auf Medikamente beobachtet werden: Bei ihnen besteht häufig "die Besonderheit ... also darin, dass es sich um eine relativ gutartige Form einer sonst schweren Epilepsie handelt". EEG-Aufzeichnungen zeigen bei ihnen häufig mehr Symmetrie und weniger Auffälligkeiten und obgleich nicht alle Kinder durch medizinische Behandlung Anfallsfreiheit erlangen, entwickeln Kinder mit Down-Syndrom im Anschluss an das West-Syndrom seltener das Lennox-Gastaut-Syndrom oder andere Formen von Epilepsie als Kinder ohne zusätzliches Erbmaterial des Chromosom 21|21. Chromosoms.
West-Syndrom bei Säuglingen mit anderen Syndromen
Neben Kindern mit Down-Syndrom (Trisomie 21) haben auch Kinder mit Bloch-Sulzberger-Syndrom, Bourneville-Pringle-Syndrom, Foix-Chavany-Marie-Syndrom und Sturge-Weber-Syndrom ein überdurchschnittlich hohes Risiko, das West-Syndrom zu entwickeln.
|